Papierloos werken
De wens om papierloos te kunnen werken wordt steeds groter, zowel binnen de ziekenhuizen als bij de CRO’s en de farmaceutische bedrijven. In de praktijk blijkt echter dat bij clinical trials papierloos werken nog nauwelijks wordt toegepast. De werkgroep richt zich de komende tijd op de mogelijkheden van papierloos werken bij klinisch onderzoek.
Elektronische Investigator Site File
Alle belangrijke documenten verkregen voor, tijdens en na de klinische studie moeten bewaard worden in een trial master file. Deze verzameling van documenten moet het mogelijk maken om na afsluiten van de klinische studie deze te kunnen valideren, de kwaliteit van de data te kunnen verifiëren en om te bewijzen dat de gehele klinische studie volgens GCP verloopt.
De trial master file is opgebouwd uit twee delen. De sponsor trial master file, alle documenten verkregen en opgeslagen door de sponsor, en de investigator site file, alle documenten verkregen en opgeslagen door de site.
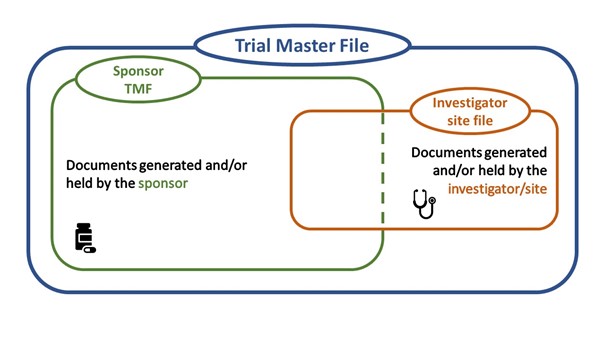
Momenteel worden nog veel investigator site files bijgehouden op een papierenversie. Echter zien we een shift om over te schakelen naar een elektronische manier van opslaan en werken. Een elektronische investigator site file brengt vele voordelen met zich mee: minder papier, minder opslagplaats nodig, makkelijker werken, het faciliteren van remote monitoring etc.
Om de verschillende sites en andere betrokkenen bij klinische studies te ondersteunen in de overschakeling van een papieren versie naar een elektronische versie of het beoordelen van het huidige elektronisch systeem heeft de DKWO-werkgroep de minimale eisen waaraan een elektronische investigator site file aan moet voldoen opgenomen in een aanbevelingsdocument.
Remote monitoring van klinische studies
Monitoring is van toepassing op al het onderzoek dat onder de reikwijdte van de WMO valt.. Veelal gebeurt deze monitoring door de deelnemende sites te bezoeken (on-site), waarbij wordt gecontroleerd of de ingevoerde data overeenkomt met de data opgenomen in de source (veelal het elektronische patiëntendossier). Dit proces noemt men source data verificatie. Het monitoren gebeurt in opdracht van de sponsor van het onderzoek. Clinical Research Oranisation (CRO) worden vaak ingehuurd om deze monitoring te verrichten.
Bij remote source data verificatie wordt de monitoring niet on-site verricht maar wordt er op afstand (off-site) ingelogd in het elektronisch patiëntendossier en de database om de ingevoerde gegevens te controleren.
De DKWO-werkgroep heeft een positiedocument opgesteld waarin het pleit voor het toestaan van remote source data verificatie en geeft in dit positiedocument een overzicht van minimale voorwaarden waaraan moet worden voldaan om rSDV te implementeren.
eConsent: praktische aanvulling op CCMO handreiking
Per 1 juli 2022 is het gebruik van eConsent binnen mensgebonden wetenschappelijk onderzoek wettelijk toegestaan. De procedure van elektronische toestemmingsverlening moet worden vastgelegd in het onderzoeksprotocol en worden beoordeeld door een toetsingscommissie. De CCMO heeft in augustus 2022 een handreiking gepubliceerd voor de beoordeling door toetsingscommissies.
De werkgroep Digitalisering Klinisch Wetenschappelijk Onderzoek (DKWO) heeft een praktische aanvulling op deze handreiking gemaakt.
Deze aanvulling geeft stapsgewijs weer wat er nodig is om elektronische toestemming op de juiste wijze te verkrijgen. Tevens worden er aandachtspunten meegegeven voor het gebruik van eConsent binnen multicenter trials en voor indieningen bij de METC.
Met deze tips en adviezen voor onderzoekers, onderzoeksgroepen en instellingen, is de wens van de werkgroep, dat het gebruik van eConsent toegankelijker zal worden binnen het onderzoekslandschap.
Het is een ‘levend document’ dat met ervaringen uit de praktijk kan worden aangepast en aangevuld waar nodig.
Voor meer informatie over de werkgroep en bovenstaande activiteiten kunt u contact opnemen via secretariaat@dcrfonline.nl.
Publicatiedatum: 13 oktober 2023
